Can We Digitize a Neuron?
From mapping the first connectome to optogenetics and engrams – exploring how scientists are learning to read, write, and simulate neurons.

There is a tiny roundworm, ~1 mm long, that thrives in rotting plant matter and compost heaps. You can barely see it with your naked eye. It's so small that it eats bacteria for breakfast. It's about the size of a comma, but much thinner, like the thickness of a human hair. Caenorhabditis elegans grows from egg to adult in about 3 days, and its whole lifespan is done and gone in under 3 weeks.
C. elegans is almost completely see-through, which means we can inspect its cells and tissues with a microscope, while still alive, and watch biology unfold in real-time with superb detail. Due to their simplicity and unique features, these humble critters constantly help us push forward the boundary of science. The Nobel Prizes in 2002, '06, '08, and '24 were awarded for discoveries made utilizing them. You could even say it has the most Nobel Prizes of any organism out there – when adjusting for brain size.
The C. elegans nervous system has only 302 neurons, which appears woefully small compared to the 100k neurons in a fruit fly's brain, let alone the ~70 million in a mouse brain, or the staggering ~86 billion in a human brain. But this small brain is compact and efficient; despite its apparent simplicity, we've discovered that C. elegans can navigate toward or away from chemicals (chemotaxis), detect and respond to mechanical stimuli (mechanosensation), experience sleep-like states, and can even demonstrate learning and memory.
In 1986, C. elegans became the first organism to have its whole connectome mapped, meaning that for the first time we could see all the neurons in an organism's brain and the synapses where they connect. The foundational paper detailing this, The Mind of a Worm, kicked off the modern study of Connectomics. Today, C. elegans is the only creature with several complete connectomes.
Even though it was 40 years ago, the science involved in that paper still sounds like science-fiction today. Researchers physically sliced worms into thousands of ultrathin (~50 nm) sections. To give a sense of scale, a nanometer is a billionth of a meter. A 50 nm slice is about the size of a small virus particle – tinier, for example, than the virus behind COVID-19. In fact, 50 nm is so impossibly small that it's smaller than the wavelength of visible light (~400–700 nm). Any details under ~200 nm are too fine for visible-light microscopes to resolve – like distant car headlights blurring into a single glow. That's why scientists needed electron microscopes to view these details, which twist and bend the boundaries of regular light microscopy with complex physics. If the width of a human hair was scaled to a football field, each of those C. elegans slices would be just under 2 inches – unbelievable!
To achieve this, worms were chemically fixed, to essentially "freeze" their cellular structures in place. Then they were put into plastic-like resin blocks so they could be cut cleanly. Scientists then used a diamond knife – yes, really – which was mounted on an ultramicrotome device. If you picture "fancy lab equipment", this device looks exactly how you'd imagine. Researchers sheared these ultra-thin slices like a high-tech deli meat cutter with the resin block advancing just tens of nanometers with each cut. (And if you'd like to splurge for a $35k+ device, you too can get charcuterie cut as thin as a virus.)
The ribbons of ultrathin sections then float on water, due to surface tension, and are picked up, commonly on copper grids or tape. Each grid went into a transmission electron microscope and electrons were shot through the ultrathin slice, creating high-contrast images of cell membranes/walls, synapses where neurons connect, and organelles ("little organs") inside the cells.
You can't lose a single section (or you could lose the neuron's connective paths), and back then, everything had to be traced by hand. Imagine a noodle smaller than a comma with 302 dots inside of it, all connected with microscopic filaments spiderwebbed throughout, and you have to slice it into thousands of discs and retrace all of the webbed connections. It took over a decade of painstaking, meticulous work.
Since this was before the advent of digital imaging and machine learning, scientists manually aligned all these sections and followed each neuron's axons, dendrites, and the synapses where they all connected. In 1986, from the 302 neurons, they counted ~5,000 chemical synaptic connections between neurons, ~600 gap junctions (electrical), and another ~2,000 neuromuscular junctions, connecting neuron-to-muscle.
Multiple worms had to be cut and imaged, since sometimes sections were lost or distorted. (It's a good job for those who enjoy 3D jigsaw puzzles with missing pieces.) But at the end of it, we got a final wiring diagram with combined data from several animals to resolve ambiguities. From 1986 until 2016, it was the only organism we had fully mapped.
We learned so many things from our first connectome: first, every neuron was identified and got its own 2-5 letter name, like "AVAR" or "SMDVL". This allowed researchers to study the function each neuron plays in behavior using loss-of-function experiments, by destroying individual neurons with precise lasers and seeing what part of behavior is affected. (This would be like intentionally breaking parts of your car to see what parts make the A/C work.) With these techniques, researchers identified specific neurons that participate in the locomotory engine of the animal, controlling forward/backward crawling (AVB and AVA), and other chemosensory and mechanosensory neurons that funnel information to these circuits. For example, there's a clear reflexive "escape response" circuit for harmful touch (ASHL/ASHR) → leading to activation of command interneurons (AVAL/AVAR) → then motor output (reverse direction and turn away).
The worm's nervous system is mostly "hardwired," but updated reconstructions published in 2014, 2016, and 2020 showed subtle variability across individuals, showing both stability and flexibility. Some connections have multiple synapses too, revealing graded strength rather than just binary links.
As we started to untangle the worm's connectome, we started to wonder: can we control the brain through manipulating the neurons we've detected? Could we somehow activate a neuron remotely, simulating a natural activation? Ultimately, could we "digitize" one of them, so that the organism still functions, even if a target neuron was destroyed?
Wildly, the answer to all of these appears to be yes.
A Mind is More than a Map
Just as the terrain of the world is more than the map we use to illustrate it, our minds are more than just the map of its neurons. For "digitizing" a neuron, there are many elements to consider – like a map that not only displays the streets, but also things like its traffic. The key measure is: "what info do we need to simulate its function?"
So what really makes up a neuron?
- Morphology - the neuron's shape; its soma (central part), and dendrites/axons (receiving and transmitting connectors).
- Connectivity - how the neurons connect; its "network map".
- Molecular identity - which genes are turned on and what molecules are present, like receptors for specific neurotransmitters.
- Electrophysiology - what it looks like when the neurons fire: how easily they fire, how fast, and what pattern; the "electrical personality".
- Biophysical models - the gold standard for fully "understanding" a neuron: distilling its morphology, connectivity, molecular identity, and electrophysiology into equations which obey the laws of physics and let us recreate the neuron's activity, as a digital twin.
Beyond just wiring and weights, we also need to understand how these properties are regulated; for example, how neuromodulators (monoamines, neuropeptides) can reconfigure effective connectivity, like during learning; we'll return to this in the proposed experiment in the appendix.
For the first two – morphology and connectivity – we discussed earlier how we can get these from electron microscopes (and nowadays, we can get many aspects from advances in regular light microscopy).
In terms of molecular identity, C. elegans is again at the forefront of scientific understanding: in 1998, it became the first multicellular organism to have its full genome sequenced. That gave us the full "reference book" of its DNA, though later work helped us see which genes are actually turned on in different cells (the transcriptome) or modified over time (the epigenome). Today, the molecular identity of every individual neuron has been established through single cell RNA sequencing. (We sure know a lot about these worms!)
The functional weights between neurons can be inferred from their structure and connectivity, but as the saying goes, correlation doesn't guarantee causation; we need some way to understand what the neurons look like when activated in response to stimuli (electrophysiology) and a framework to reproduce that (biophysical models). But the missing piece is a way we can conduct experiments by activating or inactivating individual neurons to improve our understanding. That's where the lasers come in.
Operating Worms - with Lasers
For decades, scientists have used light to activate identified neurons. This is from an amazing technique called optogenetics, developed in 2005. This "revolutionary" discovery was achieved in work by Edward Boyden, Karl Deisseroth, Peter Hegemann, and Georg Nagel, Gero Miesenböck, Ernst Bamberg, who were awarded the Brain Prize in 2013. Hailed as a transformative "method of the year" by Nature Methods in 2010, it won several awards like the Breakthrough Prize (2015), and will probably receive the Nobel Prize sooner or later. Optogenetics makes neurons electrically sensitive to light, by genetically modifying neurons with opsins – specialized light-sensitive proteins, first extracted from algae, that serve as ion channels or pumps. There are many versions of optogenetics; for example, Channelrhodopsin-2 (ChR2) opens a channel for positive ions (Na+, Ca2+) when exposed to blue light, moving voltage up in the neuron and priming it to fire. Others, like Halorhodopsin, pump chloride ions in under yellow light, making the neuron more negatively charged and silencing it.
Researchers use clever genetic tricks to get the opsins into neurons; in mammals, we often use a harmless virus to deliver an opsin gene (DNA that codes to create the opsin protein) only to specific neurons. In worms, you can directly inject the DNA into its gonads , which gets soaked up and propagated by a developing egg cell so when that single egg cell develops into an adult, that DNA is present in every cell. By adding a "regulatory promoter" (a DNA sequence that tells the transcription machinery when to start copying) to the opsin gene DNA, we can ensure the opsin is only turned on in certain cell types.
To be used, the opsin gene has to be transcribed (copied) by RNA polymerase into RNA (messenger RNA, or mRNA). This mRNA carries the instructions out of the nucleus and into the cytoplasm, where ribosomes grab the mRNA, translate the RNA code to an amino acid code with the help of tRNA, and are then strung together to form the opsin protein. After being created, they get trafficked along the subcellular microtubule highway to their final destination on the outer cell membrane, waiting for light to strike.
After the opsins are in place, then an LED, fiber optic cable, or laser can be used to shine a light, activating the protein with millisecond precision. With this, neurons can be made light-sensitive and then activated or silenced with colored light. It's miraculous.
Scientists have used optogenetics to further understand the network role of individual neurons in the worm brain. For example, activating C. elegans' ASH interneurons optogenetically makes the worm instantly crawl backward, as though it just smelled something dangerous and needs to escape. Or, if you kill the ASH neuron but then shine light on the command interneurons (AVA/AVD/AVE) normally connected to it, you can still drive the backward escape response.
C. elegans was one of the first animals where channelrhodopsins were expressed; this is aided by the worm's transparency, mapped genome, and fixed cell lineage (so we can say with confidence a specific neuron is always here, in this location, with this identity). But in recent years, scientists have taken this nifty optogenetics technique quite far indeed.
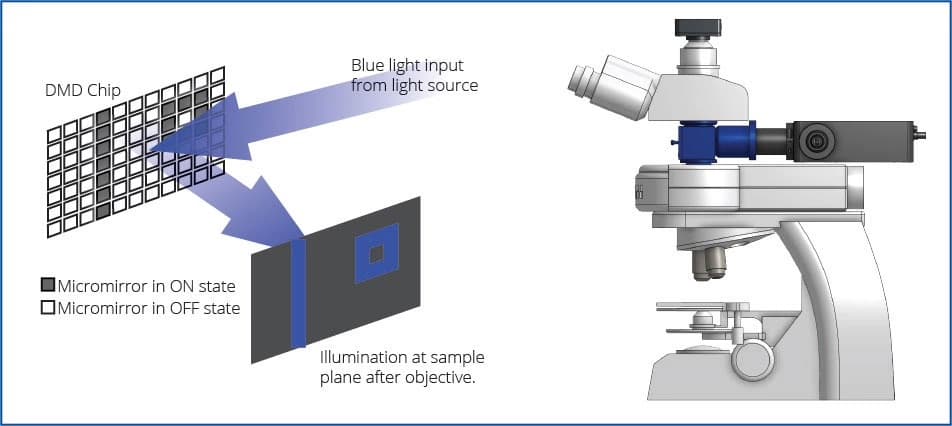
In 2011, researchers built a system called CoLBeRT that lets scientists turn individual neurons on or off in freely moving C. elegans, using light. The paper's authors genetically engineered the worm to express light-sensitive proteins in motor or touch-sensitive neurons along its body, and used a high-speed video camera (50 fps) to watch the worm as it crawled freely on a microscope stage. They wrote software that identified the worm's "body parts" as it moved to estimate where the neurons would be inside the body based on the worm's posture, and used that data to aim a digital micromirror device (DMD) which directed laser light patterns to activate or silence the neurons. The DMD can get quite accurate, down to ~30 micrometers (µm) effective resolution in moving worms; this is on the smaller end of the width of a human hair (17–181 µm on average) and allows for separately driving targets ≳50 µm apart, but they couldn't achieve single-cell precision in crowded regions of the worm's head (2–5 µm).
This feedback loop happened in real time, enabling precisely targeted stimulation of neurons as the worm wiggled around. The authors of this study were able to "push" them around with the lasers, simulating touch. But what's crazier is that researchers were able to trigger more complex (or at least interesting) behaviors, such as targeting hermaphrodite-specific neurons that normally drive egg release. Activating these actually made the worms lay eggs on command!

This type of real-time experiment is called "closed-loop" because the DMD patterning is triggered by real-time video processing. These kinds of experiments are remarkably informative for scientists, allowing for much more precise experiments. To get a sense of how precise these lasers are, check out the below GIF of UCSF neuroscientist Raymond Dunn rendering gameplay of the retro video game Sinistar on a digital micromirror device – and check out the scale in the bottom right (20 µm), showing how small some of the projected dots on the gameplay are. To be clear, this is just projecting video game footage to show the accuracy of the system – and not images of the CoLBeRT system itself.

From Worms to Flies – and Learning from our Maps
With these experiments and others, we've crossed into a territory where we can manipulate living organisms (in vivo) by activating certain neurons. Next, scientists are working on how to combine the "functional" data from neuron stimulation with the connectome. We're taking the digital maps we've made of an organism's connectome, testing them with simulations, and trying to understand how information is routed in a living organism.
In 2023, we fully mapped the first insect connectome - a larval Drosophila melanogaster, aka the fruit fly. (Fruit fly larvae are transparent, which makes it easier to deal with from an optogenetics perspective.) This represents a decade-long effort that will improve our understanding of more complex brains for decades more.
Researchers were able to manipulate the digital rendering of the brain ("in silico") to simulate real world actions and what neurons might be involved in certain behaviors. Crucially, these predictions, first found with computational analysis, were reproduced and confirmed in real living animals.
This example shows the real power of brain maps – predicting unknown things about them in ways that can be confirmed by experiment. There's still so much we don't know about brains that these maps and experiments will help us understand.
Can we Map Memories?
So far we've talked about how scientists investigate the basic building blocks of brain function, by building brain maps and stimulating neurons. But one of the most interesting things about brains is how they store and use past experiences – memories – to better interact with the world. There are several types of memory, distinguished by how they're stored and how they're incorporated into behavior. Short term memory, working memory, long term memory, and the experience of thinking and remembering – the fuzzy nature of consciousness itself.
Scientists try to take these complex concepts and break them down in specific ways so that they can be rigorously studied. In a paper published this year, it was discovered that worms have a seconds-scale short-term memory that they use to steer towards food sources. This memory is achieved by persistent temporary activity in a small interneuron subnetwork: C. elegans compares odor cues, like bacterial metabolites signaling nutritious food, across side-to-side head sweeps almost like sniffing around. The worms hold onto this alternating activity to remember which direction to turn to orient towards the food in the future. This has pointed to likely mechanisms of how the brain encodes short-term memories – via the rhythms of our brain states. By contrast, longer-lasting memories (hours to days) typically require new gene expression and protein synthesis and are stored as changes in synaptic strength and/or excitability – sometimes with structural tweaks – so they persist after moment-to-moment activity fades.
Engrams
For these longer-term memories, there's a fascinating area of research into the neurons and related cells which we believe store the critical information required for memory recall: "engrams". Even in the time of Plato and Aristotle (~350 BCE), people suspected memories are stored in enduring changes in the brain, but scientifically this field of study emerged in 1904 when Richard Semon introduced the term "engram" to describe the neural substrate for storing and recalling memories, which could be re-activated by environmental cues. Early attempts at locating engrams over the course of decades by Karl Lashley failed, and many considered it a hopeless undertaking. But over a hundred years since engrams were proposed by Semon, in 2012, Nobel Laureate Susumu Tonegawa Lab's published a remarkable study named "Optogenetic stimulation of a hippocampal engram activates fear memory recall" in Nature. The authors tagged neurons active during learning and later reactivated just those cells with light to evoke a fear memory. This built on lots of previous research:
Reijmers et al. in 2007 built the "TetTag" technique with mice to mark neurons active during learning – and showed those same cells active during recall;
Han et al. in 2009 biased specific amygdala neurons into a fear memory (by turning up a protein called CREB - basically making neurons more excitable and likely to be included in the memory formation process) – and then ablated just those cells, after which the memory could no longer be expressed.
Essentially, these researchers would train mice to associate a tone and sound with a small electrical shock, in a certain environment, known as classical fear conditioning. With tagging and optogenetics, through these studies and a series of following experiments, we now know that:
- Disrupting suspected engram neurons disrupted memory retrieval
- Re-activating them via optogenetics induced memory retrieval even without the environmental cue
- Engram cells experienced increased synaptic strength and preferential connectivity to other downstream engram cells
- We can manipulate memory encoding to generate false memories – or even "implant" some – such as making a mouse "scared" of a new environment, where no shock had occurred in the past
- And "silent" engrams were discovered in amnesic mice, where re-activating them via optogenetics induced retrieval
We have slowly started to peel away the mystery of memory, poking and prodding at the edges, to understand the underlying mechanisms.
Can we Digitize Memories?
An interesting experiment would be to suppress the expression of an engram by silencing critical neurons – to make it "forget" – and then try to simulate the behavior again by activating the necessary downstream circuit elements, bypassing the engram cells completely. This could make an animal act like it remembered the electric shock, even without engram cell activation. But to claim we've digitized a memory, we'd need more than a wiring diagram. We would also need data from experiments stimulating neurons, persistent activity states, and a whole lot else relevant to brain function (e.g. neuromodulators). This is detailed further in the appendix.
As this field inches forward, we may find that more of our organic computations can be digitized – raising profound questions for us humans in the long run.
Can this be Done in Humans?
Although many human genes have a counterpart in worms (about 40%!), humans are a bit different from C. elegans. For one, we have skulls. We're not so transparent. To get a sense of scale, if you had a grain of rice for every neuron in a brain, the rice from our friendly roundworm could fit in your hands, about 6.5 grams worth, hardly a few bites. But a human brain's worth of rice would take up almost 60 freight trucks. These techniques can translate to other organisms in general, but the leap to a human brain is enormous due to mechanical things like scale and tissue opacity, but also regulatory and ethical considerations. Digitizing a neuron in C. elegans compared to one in a human brain is a bit like comparing swimming a lap in your local pool with crossing the Pacific – the human brain (and skull) are formidable opponents in this realm. But there are promising ways we can tackle this problem – ranging from invasive electrode arrays put directly into brains, such as with Elon Musk's Neuralink device (it sounds Cyberpunk but there are many companies trying this), or minimally invasive ones that can be inserted into the brain like stents within veins such as Synchron. In 2015 we also advanced the techniques of optogenetics so that we can achieve similar results via transcranial focused functional ultrasound (tFUS) and mechano-sensitive channels and reporters, instead of using light-activated optogenetics which is challenging to use with our dense skulls and opaque, voluminous brains.
In the next post in this series, we'll explore this wild frontier and all the techniques we have at the present for Brain-Computer Interfaces (BCIs), how we can read/write with them, and the case for how it might be done in the future – a future which is closer than you might think!
Appendix
Electron Microscopes
Unlike light microscopy, which works by shining a light through a sample and using glass lenses to bend and focus the light so the image looks bigger to your eyes (or a camera), like a magnifying glass, electron microscopes take pictures using a beam of electrons.
Light waves can only get so small - about 400 nm (violet) to 700 nm (red) - and things smaller than ~200 nm, like viruses, can't be resolved. (Although there are some new tricks with light microscopes; see below.)
Electrons are quantum particles; in the microscopes we use, as we accelerate them, their wavelengths can get extremely tiny – often .005-.05 nm – so they can resolve images on the order of .1 nm! With this, you can see actual atoms; at that scale, over a million of them could fit lined up across the width of a human hair.

Electron microscopes are so sensitive that they have to be in a vacuum so the electrons don't bump into air. Powerful magnets act like lenses to focus the electrons, and we can monitor where they pass through or hit the sample, which we can resolve into an image.
Advances in Light Microscopy
Electron microscopes are incredibly powerful, but expensive and complex. Clever new techniques have been developed that allow us to use conventional microscopes, yet at drastically better resolution, such as Expansion Microscopy (ExM). In 2015, ExM was introduced by Chen, Tillberg, and Boyden in a paper in Science , which allows us to get resolution down to the ~70 nm range (or ~20 nm with iterations) via physically swelling the sample after embedding it in a polymer, expanding it in a uniform direction. (It's not too far from the type of polymer seen in baby diapers that swells when in contact with liquid!) This is very powerful for large-scale mapping (ideal for connectomics), and it can be combined with other techniques. For example, with LICONN (Light-microscopy-based connectomics), developed by scientists at the Institute of Science and Technology Austria (ISTA) and Google Research, researchers combine this hydrogel expansion that expands cells up to 16x – without breaking the connections between neurons – which help us to resolve the densely packed neural structures like axons and synapses. Proteins are also stained with fluorescent tags to highlight specific molecules, giving us a much richer picture than we can get with electron microscopy (which would only show physical structures, not molecular makeup). And with recent advances in AI, we can now use "flood filling" algorithms and similar programs to better automate tracing of connections and processing of these images. It's fast, too, and can be applied to larger volumes of tissue compared to EM, which has made far-off targets such as mapping the first full mouse brain (and later, hopefully, human brain) within reach in the coming decade or so.
One limitation is that it can't do live imaging, but other techniques have been developed for that, too, which leverage modern advanced computing hardware to solve longstanding problems in optics. One example is Structured Illumination Microscopy (SIM), which projects patterned light (like stripes) onto the sample and uses the interference (moiré patterns) to computationally reconstruct higher-resolution information. This gets us to about 2x better than the normal diffraction limit while still working with live cells.
"Digitize a Memory": A potential research project in mice
Aim: Show that you can bypass an engram node and still retrieve the memory by reinstating the right downstream network state (via a "digital twin"), and that this is more than momentary mimicry.
Bonus: This could also be paired with a sonogenetic approach (vs. optogenetics) to mirror human-compatible technologies that may be used in Brain-Computer Interfaces over the next decade.
Steps (summarized for brevity):
-
Train a canonical memory in mice either via contextual/environmental fear or a tone-shock fear, mirroring past engram studies (and keep matched controls: no shock, or unpaired tone/shock). Classical fear conditioning is exceptionally well studied and (relatively) ethologically relevant.
-
Tag the ensemble of cells and record the network state
- Activity-dependent tagging : during learning, open a brief window so neurons that are active (e.g. immediate-early gene promoter logic) express:
- A reporter (fluorescent protein to verify tagging), and
- A sonogenetic actuator (an ultrasound-responsive ion channel) in the targeted region (e.g. DG/CA1 for context; BLA for the subjective feeling, or "valence").
- Result: only the neurons that were active during a short time window during learning will express the actuator gene
- Record the population dynamics of neurons during recall, for example via 2-photon, miniscope, or Neuropixels (hippocampus <> amygdala <> mPFC) . Neuropixels has the advantage for measuring Local Field Potential (LFP) for evaluating theta-phase timing – a rhythmic oscillation in the hippocampus that organizes when different regions talk to each other. However the tradeoff of spatial resolution with Neuropixels i.e. getting sufficiently close to engram cells is an open challenge.
- Compress to "state variables": fit a low-dimensional state vector (spatial cell pattern + oscillation phase features, depending on optophysiology/electrophysiological approach) that best predicts successful recall (e.g. freezing onset/duration).
- Activity-dependent tagging : during learning, open a brief window so neurons that are active (e.g. immediate-early gene promoter logic) express:
-
Test causality with sonogenetic read-write
- Necessity (silencing upstream node) – reversibly turn down the tagged upstream ensemble during recall using ultrasound either via:
- Interneuron route: express the sonogenetic actuator in the local inhibitory interneurons (e.g. in DG or BLA). Brief tFUS bursts should enforce a net inhibition of the engram ensemble.
- Inducible expression route: use an inducible actuator in the engram itself with parameters that produce reduced excitability during recall. (Prediction: natural recall drops.)
- "Digital twin" sufficiency (bypass via downstream state) – while the upstream engram is silenced, deliver a patterned tFUS sequence to the downstream target (e.g. CA1 or BLA) that recreates the recorded recall state (cell-set targeting + theta-locked timing). Prediction: recall appears without the upstream node – i.e. state reinstatement drives the behavior.
- BCI alignment: Replacing optical holography/DMD from past experiments with phased-array tFUS patterns.
- Necessity (silencing upstream node) – reversibly turn down the tagged upstream ensemble during recall using ultrasound either via:
-
Separate mimicry from a digitized memory
- Mimicry (weak): The behavior only exists while tFUS is on and collapses immediately when stopped.
- Digitized state (strong): After suppressing natural expression, a brief digital-twin replay restores recall with appropriate persistence (seconds-scale after-effect) and cue specificity. In other words, no freezing in a neutral context.
-
Add the chemical context if needed: If 4b fails, measure neuromodulators during successful recall (e.g., NE/ACh/DA sensors).
- Re-run replay with vs. without the matching neuromodulatory context (e.g., sonogenetically nudge locus coeruleus for NE or medial septum for ACh).
- Inference: If replay only works in the right chemical mode, that chemistry is part of the digitized state.
-
Rescue (strong claim)
- Keep the upstream pathway silenced, then restore the behavior by driving the downstream digital twin (+ required chemistry from Step 5).
- Bonus: demonstrate bidirectionality – use a complementary pattern to dampen recall. Beyond demonstrating control of the system, this would directly imply a therapeutic intervention target.
Controls & practical notes (ultrasound-specific)
- Shams & scrambles: sham ultrasound controls (test doing everything the same but don't actually pulse); scrambled cell sets and time-shuffled phases (test playing wrong pattern/timing); non-expressing animals (test without the sonogenetic channel).
- Auditory/thermal controls: control for cochlear confounds and heat (audible artifacts, thermometry).
- Targeting: neuronavigation; skull model-based focusing; verify focal spot with imaging/physiology.
- Closed-loop: gate stimulation by EEG/LFP phase and behavior, as you would in a human closed-loop BCI.
- Whole-brain check: post-hoc activity mapping to confirm the digital-twin recruits the same broader network as natural recall.
This hypothetical experiment above is both completely inadequate for a real research proposal, yet too full of jargon for the average layperson to understand. But conceptually it's simple: read the same inputs, compute the same transform, write the same outputs, then see if memory makes it through. Why build a research programme like this? If it works, we push forward what we know about memories, and potentially untether them from our physical brains – plus line up the required equipment with what we know will be possible for human BCIs in the medium/long-term. We live in interesting times!
Acknowledgements
Massive thanks to Ray Dunn for reading and editing this post, and for making it actually scientifically literate. He is possibly one of the few people who appreciates C. elegans more than I do!
For corrections or inquiries, please contact
corbett@these.us